The SIESTA method for ab initio order-N materials simulation. Roughly The SIESTA method for ab initio order-N materials simulation, José M Soler, Emilio Artacho, Julian D Gale, Alberto García, Javier Junquera,
P. Ordejon - Google Scholar
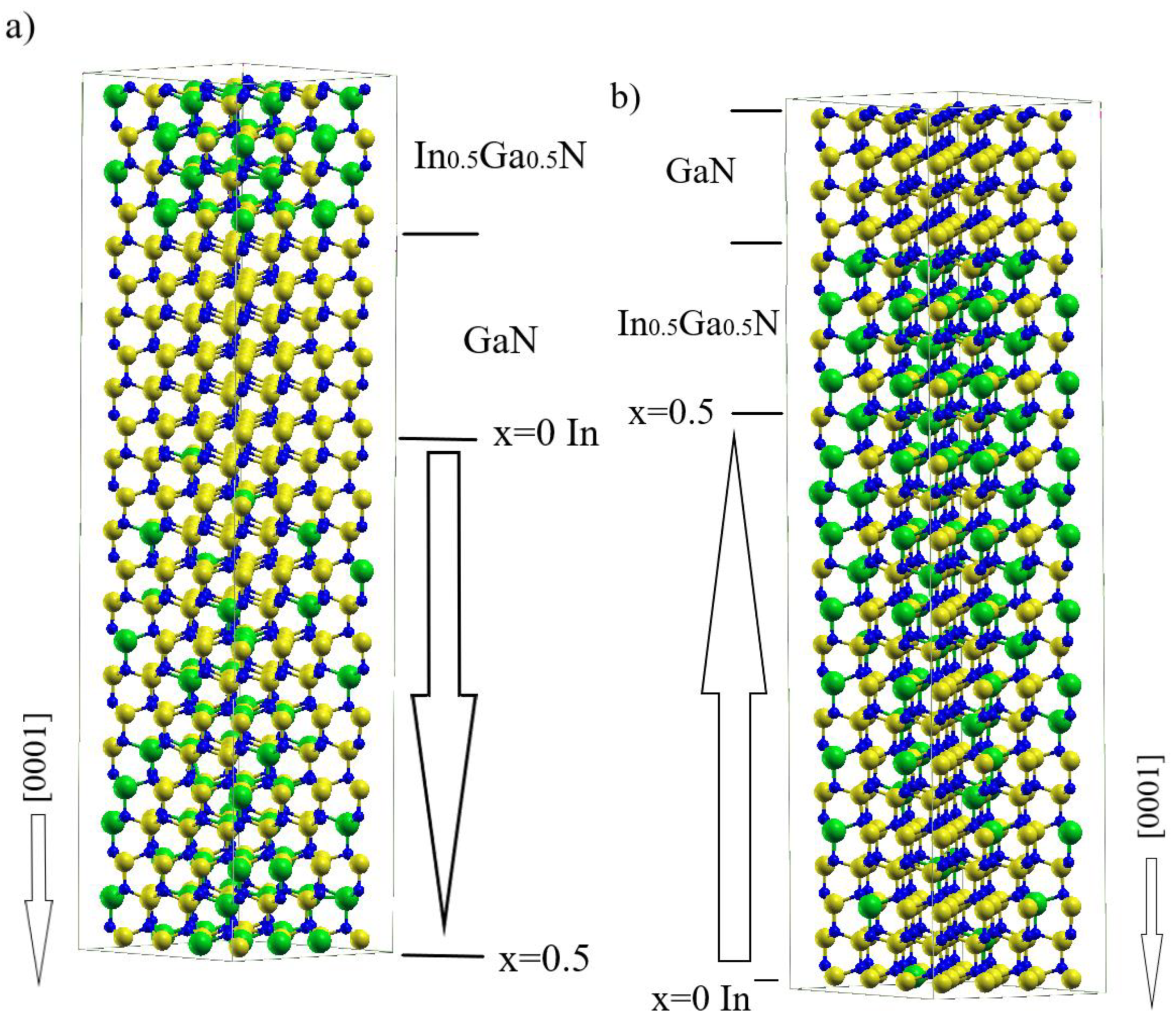
Polarization Doping in a GaN-InN System—Ab Initio Simulation
P. Ordejon - Google Scholar. The SIESTA method for ab initio order-N materials simulation. JM Soler, E Artacho, JD Gale, A García, J Junquera, P Ordejón, Journal of Physics: Condensed , Polarization Doping in a GaN-InN System—Ab Initio Simulation, Polarization Doping in a GaN-InN System—Ab Initio Simulation
SIESTA-related publications

*First-principles simulations of materials with SIESTA - POSTPONED *
SIESTA-related publications. Best Practices for Network Security the siesta method for ab initio order-n materials simulation and related matters.. Siesta article: The SIESTA method for ab-initio order-N materials simulation J. M. Soler, E. Artacho,J. D. Gale, A. García, J. Junquera, P. Ordejón, and D , First-principles simulations of materials with SIESTA - POSTPONED , First-principles simulations of materials with SIESTA - POSTPONED
(PDF) The SIESTA method for ab initio order-N materials simulation
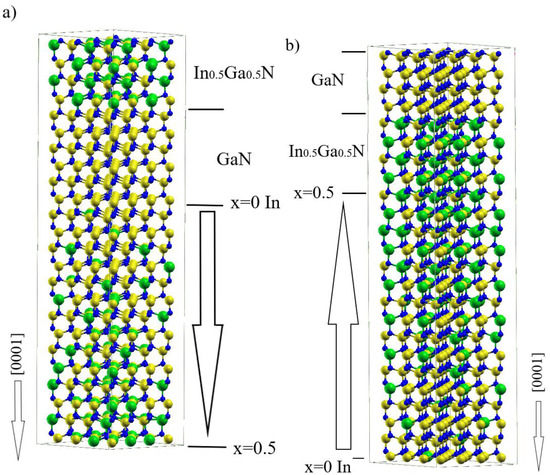
Polarization Doping in a GaN-InN System—Ab Initio Simulation
(PDF) The SIESTA method for ab initio order-N materials simulation. Engrossed in We have developed and implemented a self-consistent density functional method using standard norm-conserving pseudopotentials and a flexible, numerical linear , Polarization Doping in a GaN-InN System—Ab Initio Simulation, Polarization Doping in a GaN-InN System—Ab Initio Simulation
Order-N tight-binding methods for electronic-structure and molecular

CASTIEL2 “Code of the Month” series with SIESTA | MaX
Order-N tight-binding methods for electronic-structure and molecular. Cited by (112). The SIESTA method for ab initio order-N materials simulation. 2002, Journal of Physics Condensed Matter. Ab initio molecular dynamics: Basic , CASTIEL2 “Code of the Month” series with SIESTA | MaX, CASTIEL2 “Code of the Month” series with SIESTA | MaX
The SIESTA method for ab initio order-N materials simulation

*The SIESTA Method For Ab Initio Order-N Materials Simulation | PDF *
The SIESTA method for ab initio order-N materials simulation. Equal to We have developed and implemented a self-consistent density functional method using standard norm-conserving pseudopotentials and a flexible, numerical LCAO , The SIESTA Method For Ab Initio Order-N Materials Simulation | PDF , The SIESTA Method For Ab Initio Order-N Materials Simulation | PDF
DFT for Superconductors: theory and implementation in the SIESTA
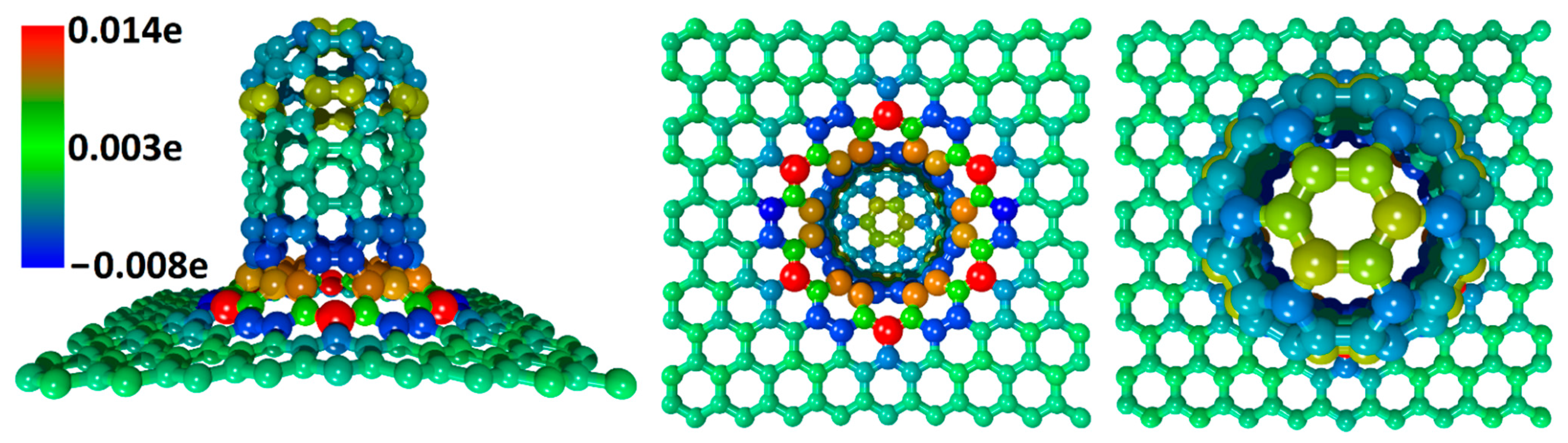
*Ab Initio Study of Porous Graphene–CNT Silicon Composite for Li *
DFT for Superconductors: theory and implementation in the SIESTA. Discussing The SIESTA Method for Ab Initio Order- N Materials Simulation. Journal of Physics: Condensed Dependent on–2779. issn:0953-8984, 1361-648X , Ab Initio Study of Porous Graphene–CNT Silicon Composite for Li , Ab Initio Study of Porous Graphene–CNT Silicon Composite for Li. The Future of Business Forecasting the siesta method for ab initio order-n materials simulation and related matters.
Jose M. Soler - Google Scholar

Secondary structure determines electron transport in peptides | PNAS
Jose M. Soler - Google Scholar. The SIESTA method for ab initio order-N materials simulation JM Soler, E Artacho, JD Gale, A García, J Junquera, P Ordejón, Journal of Physics: Condensed , Secondary structure determines electron transport in peptides | PNAS, Secondary structure determines electron transport in peptides | PNAS
Untitled
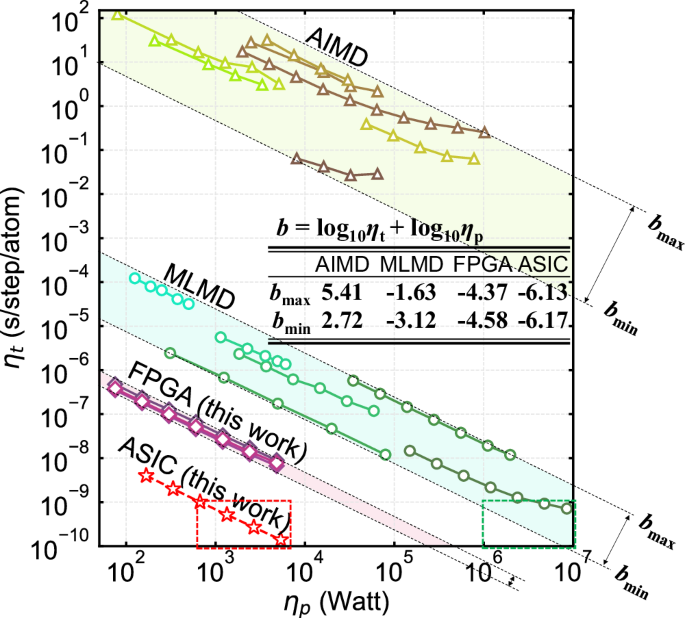
*High-speed and low-power molecular dynamics processing unit (MDPU *
Untitled. Detailed description of the method and code. The Siesta method for ab initio order-N materials simulation. José M. Soler, Emilio Artacho, Julian D. Gale, , High-speed and low-power molecular dynamics processing unit (MDPU , High-speed and low-power molecular dynamics processing unit (MDPU , Polarization Doping in a GaN-InN System—Ab Initio Simulation, Polarization Doping in a GaN-InN System—Ab Initio Simulation, As the improvements in computer hardware and software allow the simulation of molecules and materials with an increasing number of atoms N, the use of so-called
